

1. 서 론
본 연구는 완제 D-항원 정량 시험법의 유효성과 동등성 검증하는 방법을 의약품 설계기반 품질고도화(Quality by Design, QbD)6시그마® 프로세스로 수행하여 단일변수시험(One Factor At a Time, OFAT)기반의 시험법 밸리데이션과 시험 결과 허용 기준과 단순 비교했던 방법에서 분석법에 적용되는 의약품품질고도화활동(Analytical Quality by Design, AQbD)기반의 시험법 밸리데이션과 시험 결과를 통계 분석하여 기존의 방법보다 허가자료의 신뢰성을 높여 허가 변경 지연 리스크를 감소시키는 방법에 대한 연구이다. 기존에는 국제의약품규제조화위원회(International Council for Harmonisation of Technical Requirements for Pharmaceutical for Human Use, ICH) Q2에 따른 시험법 밸리데이션은 OFAT방식이고, 시험법 변경 전후에 D-항원 함량 동등성을 비율로서 확인하였는데, 이를 AQbD기반으로 수행하였다. 시험방법 밸리데이션에 AQbD 개념을 도입하여 고위험 요인을 실험계획법을 통해 교호작용을 확인하고, 위험평가(Risk Assessment, RA)를 통해 고위험 요인의 단독/복합 작용을 검증하여 시험방법의 성능의 항상성을 유지할 수 있고, 시험법 변경 전 후의 D-항원 함량 동등성을 통계적인 방법으로 입증하여 시험방법 밸리데이션시 재시험율 감소를 기대할 수 있었다.
OFAT으로 시험법을 검증했던 과거의 방법은 한 번에 하나의 변수들을 변화시켜 가며 확인하였기 때문에 시간도 많이 소요되고, 하나의 변수들의 선형관계만을 확인하고, 변수들 간의 교호작용(Interaction)을 확인하지 못했다. 시험방법 밸리데이션시 검증 실패에 대한 주요 원인이었다. 이를 보완하여 본 연구는 QbD6시그마 프로세스를 도입하여 시험방법의 성능을 높이고, 항상성을 유지할 수 있는 방법을 제안하였다. QbD6시그마 Define단계에서는 분석법의 목표(Analytical Target Profile, ATP)와 핵심시험방법 요소(Critical Method Attribute, CMA)의 목표를 정의하였고, Measure단계에서는 측정시스템 분석, Analyze단계에서는 위험평가(Risk Assessment, RA)를 통해 CMA에 영향을 주는 핵심방법 요소(Critical Method Paremeter, CMP)를 선정하고, Improve단계에서는 실험계획법을 통해 시험방법 밸리데이션과 동등성을 검증하였고, Control 단계에서는 일탈(Out Of Target, OOT)의 모니터링 계획을 수립하고 CMP에 대한 관리전략(Control Strategy, CS)를 수립하였다(Kim and Kim, 2022).
D-항원 정량에 사용하는 항체에 대해 항체를 제조하여 이를 시험할 시험법 개발이 필요한 경우 식약처 등 기관에 제출할 허가자료 작성이 필요하고, 신규 시험법 밸리데이션과 기존시험법과 신규 시험법의 동등성을 입증해야 한다. 자료가 미흡할 경우 허가 지연이 되어 개발 기간 및 매출에 영향을 줄 수 있다. ICH Q2에 따른 시험방법 밸리데이션은 OFAT방식이고, 시험법 변경 전, 후에 대한 D-항원 함량 동등성을 비율로 확인하였는데, 이를 AQbD기반의 시험방법 밸리데이션을 위한 QbD6시그마 프로세스대로 수행하였고, 실험계획법과 위험평가기법을 활용한 사례를 제시하였다. 시험법 변경 전, 후에 대한 D-항원 함량 동등성을 통계적으로 입증하여 허가 변경 자료의 타당성과 신뢰성이 향상되었다(Kim and Rhee, 2017).
QbD는 1992년 Joseph M. Juran 박사가 제안하였다. AQbD는 QbD와 동일한 원칙의 분석 방법의 개발에 적용되었고, 결과가 잘 이해되도록 목적에 적합하게 의약품의 수명 주기 전반에 걸쳐 의도한 성능을 일관되게 유지할 수 있도록 제시한 방법론이다(Kim and Kim, 2022). 프로세스에서 얻은 광범위한 지식은 적절한 분석법 성능을 제공하는 분석설계영역(Method Operable Design Region, MODR)을 설정하는 데 사용된다.
QbD6시그마는 제약업에 필수로 적용이 되어야 하는 QbD를 6시그마 프로젝트로 수행할 수 있도록 고안한 방식이다. D-항원은 바이러스의 활성을 나타내는 항원으로 항체 형성(면역원성)에 가장 중요한 인자이다. 따라서 백신 내 D-항원 함량을 품질관리항목으로 규격을 설정하여 관리하고 있다. D-항원 정량에 사용되는 항체를 제조하여 이를 이용한 신규 시험방법을 개발하는 과정을 QbD6시그마 프로세스로 소개한다.
2. 이론적 배경 및 선행연구
2.1 ICH
ICH는 의약품 허가를 위한 국가별 요구사항이 상이하여 허가 과정에서 불필요한 비용과 기간이 소요됨으로 인해 의약품의 가격 상승 및 공급 지연으로 인한 환자들의 불이익이 되는 문제를 해결하고자 1990년 미국, 유럽연합(EU), 일본의 주도로 결성되었다. 이 기구의 목적은 의약품 허가 관련 요구사항의 국제적 조율이고, 회원국들은 협의를 통해 가이드라인을 제시하고 자국의 의약품 허가 요구사항으로 채택한다. 우리나라는 2016년 11월에 6번째 회원국으로 가입되어 있다(Kim and Rhee, 2017).
의약품 개발 및 품질 관리의 기존 방식에서 탈피해 설계 및 개발 단계에서부터 제품과 공정에 대한 깊은 이해와 철저한 공정관리를 통해 의약품의 불량이 발생되지 않도록 하자는 취지의 QbD와 AQbD는 ICH에서 가이드하고 있다(Kim and Rhee, 2017).
ICH 가이드라인은 4가지로 분류되고 있다. Q가이드는 Quality Guideline이다. Q8부터 Q11까지에 QbD와 AQbD에 관련한 가이드가 제시되어 있다. ICH Q11은 API합성 공정 개발을 위한 QbD 접근방식을 설명하였고, AQbD 분석 개발 방법에서 QbD접근 방식을 따를 수 있다. Table 1은 ICH 가이드라인에서 QbD와 AQbD에 관련된 Q8, Q9, Q10, Q11에 대하여 핵심적으로 다루는 사항에 대해 요약하였다.
2.2 QbD와 AQbD 절차
QbD는 의약품의 전반적인 설계에 대한 개발관점이고, AQbD는 의약품 개발에 필수적으로 적용해야 하는 시험방법 밸리데이션을 과학적으로 수행하는 절차를 ICH가 가이드하였다(Helen, 2013). Table 2는 QbD와 AQbD의 절차를 비교하였다.
QbD는 품질목표사항(Quality Target Product Profile, QTPP)을 정의함으로 과제가 시작되고, QTPP에 영향을 주는 핵심품질특성(Critical Quality Attribute, CQA)을 RA를 통해 도출한다. 도출된 CQA에 영향을 주는 핵심공정변수(Critical Process Parameter, CPP)를 통계적인 수법을 통해 검증하여 찾고, CPP의 허용범위라고 할 수 있는 설계공간(Design Space, DS)을 도출한다. 이때 실험계획법 중 반응표면분석법이 사용된다. CPP의 관리전략을 수립하고 공정을 개발하고 생산에 돌입한다(Kim and Rhee, 2018).
AQbD는 ATP를 정의함으로 과제가 시작된다. ATP에 영향을 주는 CMA는 RA를 통해 도출한다. 도출된 CMA에 영향을 주는 CMP를 통계적 수법을 통해 검증하여 찾고, CMP의 허용범위인 MODR을 실험계획법 중 반응표면분석법을 사용하여 도출한다(George, 2013).
QbD에서 QTPP는 임상관점과 환자관점의 품질목표사항이고, CQA는 개발자 관점에서의 QTPP를 달성했는지 여부를 판단할 수 있는 정량적지표이다. CPP는 CQA에 치명적인 영향을 주는 공정변수이고, 과학적인 방법으로 도출해야 한다. DS는 설계공간의 개념으로 하나의 점의 형태인 값이 아니라, 범위를 설정하여 CPP의 허용범위를 의미한다. 그리고 CS를 설정하고, 이때는 CPP로 선정된 모든 변수를 고려해야 한다. AQbD에서는 시험법 혹은 시험분석법을 개발하고 검증하는 것이 목적이다. 이에 분석법의 목표인 ATP를 선정한다. 그리고 핵심시험방법요소 CMA를 선정한다. CMA는 ATP를 달성했는지 여부를 파악할 수 있도록 선정이 되어야 한다. RA를 통해 CMA에 영향을 주는 CMP를 선정하고, 이 때는 과학적인 방법을 반드시 사용해야 하고, DOE나 회귀분석 등을 많이 사용해서 검증을 한다. 이후에 검증된 CMP에 대한 CS를 설정하여 시험법 밸리데이션을 완료하게 된다(Kim and Kim, 2022).
QbD와 AQbD 모두 연속적으로 모니터링하여 설계된 대로 잘 진행되는지 지속적으로 검증하고, 필요 시에는 개선활동을 신속하게 진행한다. QbD가 의약품 개발 전반의 프로세스라면 AQbD는 의약품 개발 시 필요한 시험법의 검증을 위한 프로세스이다(Kim and Rhee, 2018).
3. QbD6시그마
본 논문에서는 선행연구를 바탕으로 D-항원 정량 시험법의 유효성과 동등성에 관한 연구를 QbD6시그마 프로세스로 진행하여 기존시험법과 신규시험법 간 D-항원 함량 동등성을 통계적으로 검증하였다.
3.1 Define
전체 과제의 흐름과 전개도를 Y-y전개를 통해 확인하고, Product에 대한 CMA별로 개선과제 활동을 특성요인도(Logic Tree)로 서술했다. ATP를 수립하고, D-항원 함량 시험의 유효성 즉 정밀성, 정확도, 범위를 검증하고 동등성(Equivalence) 검증 계획을 포함하여 Table 3과 같이 정의하였다.
Table 4와 같이 ICH Q2(R1)및 USP에 가이드에 의하여 CMA의 분류 항목 중 정밀성(Precision)은 전시스템 변동(Systemic Variability), 정확도는 고유무작위 변동(Inherent Random Variability) 항목으로 ICH에서 정의하며, 정량 시험(Assay for Content)에 고려되어야 할 인자이다. 범위(Range)는 배치 단위의 안정성(Stability) 목적 시험이므로 규격과 목표(Target) 농도 범위를 포함해야 한다. 비교가능성(Comparability)은 주 검증(Primary Validation)뿐만 아니라 시험법 변경에 따른 비교 검증(Comparative Validation) 수행도 필요하다.
기존의 방법은 과거의 경험으로 시험법에 영향을 주는 항목을 선정했다면 제안하는 방법은 위험평가를 통해 시험법에 영향을 주는 핵심적인 특성을 CMA로 정의하고 CMA의 Category를 선정하고, CMA의 대상특성에 대해 규격과 목표를 CMA의 대상특성(Target Characteristics)의 기준 선정을 진행한다. 본 논문에서는 실 사례를 바탕으로 수행하였고, Table 4와 같은 기준 선정은 정밀도와 정확도 비교가능성의 시험법 편차 20%를 고려하여 설정하였다. 범위는 규격 하한(60 DU/mL)과 목표농도(100 DU/mL)에서 ±20%를 고려하여 4.8 DU/mL, 12.0 DU/mL로 설정하였다. D-항원 정량 시험법은 연속 희석한 표준품과 검체의 농도-반응 곡선을 비교하여 검체의 상대 역가(Relative Potency)를 구하는 생물검정(Bioassay)이다. 절대 함량이 아닌 비율로 계산되는 상대 함량이므로 백분율 상대 편향(Percent Relative Bias, %RB) 단위를 사용하였다. 표준품(S) 대비 검체(T)의 상대 함량(ρ)과 흡광도는 Figure 1과 같이 로그/지수 관계로 증감하는 Antilog 함수로 표현된다. 로그로 변환해야만 정규 분포를 따르므로 로그 변환하여 데이터를 전처리하여 기하변동계수(Geometric Coefficient of Variation; GCV) 단위를 사용하였다(Seo and Byun, 2021).
3.2 Measure
D 항원 함량 시험의 유효성과 동등성을 기존의 방법에서 개선 방법으로 실시하였다. Table 5에서 시험방법 밸리데이션의 새로운 방법 활용 내용을 요약하였다. 과거의 방법은 한 명의 실험자가 6번을 반복하여 정확도를 보고, 1명의 실험자가 3번을 반복하여 재현성을 보았는데, 2명의 실험자가 2개의 정확도와 정밀도를 위해 동시에 실험을 하였다는 점과 단일 변수 시험이라는 방식으로 한 번에 하나의 변수를 조정해 가며 실험을 했다면, 제안한 방식은 실험계획법을 통한 분석을 통해 주효과와 OFAT으로는 알 수 없는 교호작용을 알게 되어 보다 정확하고 정밀한 시험법을 만들게 되었다는 점. 과거에는 변경 전 후의 차이만을 볼 수 있었다면 지금은 그 차이와 동등성까지도 검증이 된다는 점이 기존 방법과 다른 특징을 가지며 우수하다.
3.3 Analyze
전통적인 방법은 과거의 경험으로 CMA에 영향을 주는 CMP를 선정했다. 본 논문에서는 RA를 통한 CMP선정 방법에 대해 설명한다. 시험의 주요 단계를 프로세스 맵을 통해 메인 프로세스와 서브 프로세스로 매핑하였다. 프로세스도(Process Map)과 특성요인도(Fishbone Diagram)를 통해 확인한 ‘프로세스 입력’을 나열하고 고장모드영향분석을 통해 위험을 정량적으로 평가하였다. 여기서 위험우선순위(Risk Priority Number, RPN)는 심각도(Severity) x 발생도(Occurrence) x 검출도(Detectability)를 계산한 값이다. CMA에 영향을 주는 CMP후보가 심각도 측면에서 영향이 크고(score=4), 발생 빈도는 중간이며(score = 3), 검출하기는 어려움(score = 5)이라고 나온 경우 PRN은 4 x 3 x 5 =60이다(Cho and Lee, 2022).
심각도는 입력통제(Input Control) 실패가 CMA에 미치는 영향이 미미함(score=1)에서 큼(score=5)의 범위로 점수를 주고, 발생도는 입력 통제 실패가 발생하는 빈도가 낮음(score=1)에서 높음(score=5)의 범위로 점수를 주고, 검출도는 실패의 검출이 쉬움(score=1)에서 어려움(score=5)의 범위로 위험에 대한 점수를 주어 CMP후보를 선정한다. 이 연구에는 RPN이 20을 초과하는 경우를 CMP후보로 선정하였다.
3.4 Improve
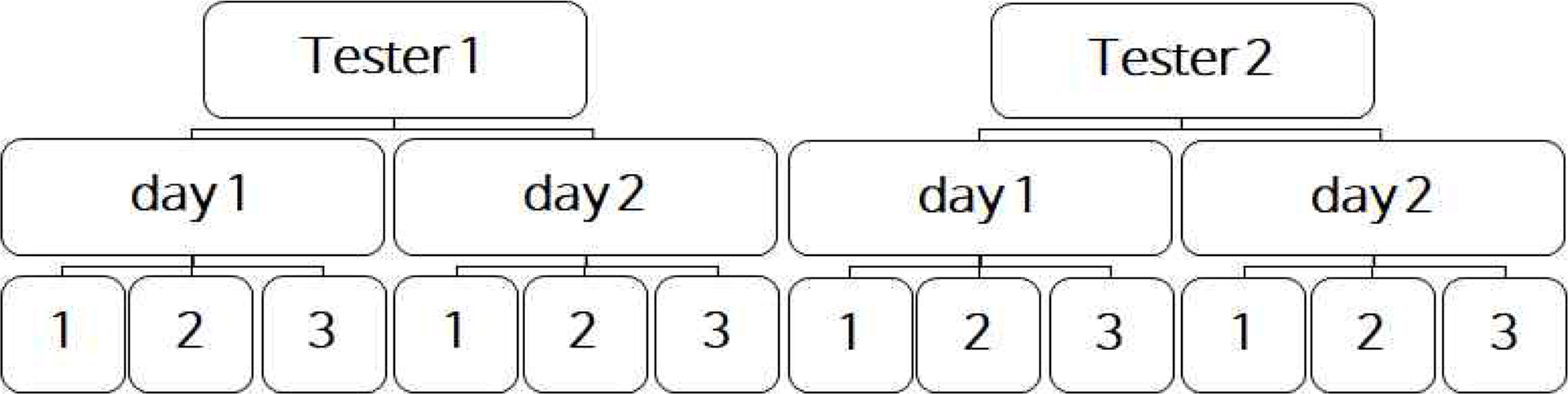
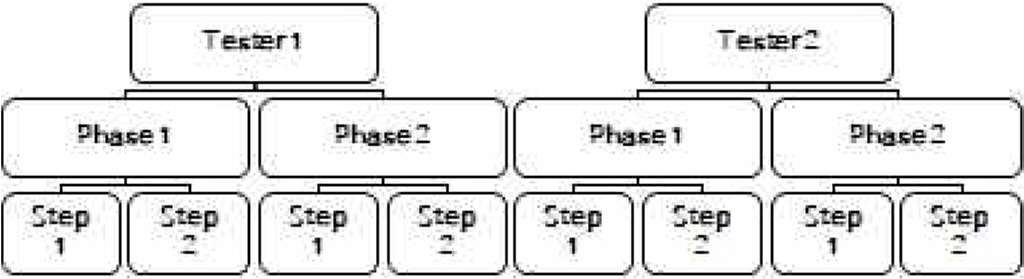
정밀도 실험설계를 CMP 중 분석가(Analyst), 실험일자(Day)의 변화에 따른 변동성을 정밀도 항목으로 평가하였다. 시험자1과 시험자2가 동일한 디자인(Balanced Design)으로 시험하여 분산 분석을 통한 분산성분분석(Variance Component Analysis)이 가능하도록 하였다.
정확도 실험결과 분석은 미국약전(US Pharmacopeia, USP)1033의 상대정확도(Relative Accuracy) 항목에 따라 시험을 실시한다. 분석가 1(Analyst I), 분석가 2(Analyst Ⅱ)가 동일한 디자인으로 각 시험자가 3회씩 측정하였다. 로그로 변환하였을 때 등간격이 되도록 검체를 제조한다. 측정값을 자연로그로 변환하고, 로그변환 값의 평균과 표준편차 90% 신뢰구간을 계산한다. 역 로그를 취하여 값을 환원하여 USP1033의 계산식에 따라 %RB(Relative Bias)를 계산한다. 정확도 실험의 결론으로 이론값(Nominal Potency) 대비 측정값(Observed Potency)의 상대적인 편차(%Relative Bias)는 -2.1 ~ 2.3 %로 허용 기준(-17%≤%RB≤20%)을 만족함을 확인하였고, 5가지 농도(36 ~ 141% Potency level)에서 90% 신뢰 구간이 0을 포함하고 상한과 하한이 허용 기준 내이므로 농도(Potency level)에 따른 편의 경향(Bias Trend)은 없음을 확인하였다.
정밀도 실험결과 분석은 측정값을 자연로그 변환하고 일원분산분석으로 분산값을 확인한다. USP1033의 3.1항 기준에 따라 실험의 변동 Var(Run)과 실험오차의 변동 Var(Error)을 계산한다. Run의 분산과 Error의 분산 값을 확인한다. Var(Error)는 군내변동(Within-run Component간 Variability)으로 MSE(Mean Square Error)로 계산할 수 있다. Var(Run)은 군간변동(Between-run Component간 Variability)과 같다. MS(Run)은 Var(Error)+ Replicates(n)*Var(Run)이고, 각 시험자가 3회씩 측정하여 시험을 실시하였기 때문에 Replicates(n)은 3이므로 Var(Run)로 정리하면 (MS(Run) – Var(Error))/3이 된다. USP1033내 계산식에 따라 중간정밀도(Intermediate Precision, IP)를 계산한다. 결론으로 군내변동과 군간변동을 모두 고려하여 IP를 계산한 결과를 확인할 수 있다. Figure 3과 같이 IP는 변동계수(Coefficient of Variation, CV)로 구하고, 표준편차(σ)를 평균(μ)으로 나눈 값이다.
견고성(Robustness) 실험결과 분석은 실험계획법을 이용하여 2수준 완전요인배치를 수행한다. 인자는 항체별로 3인자로 선정하고, 반응값도 농도별로 3개 값을 측정하여 입력한다. 실험결과 값을 자연로그 변환하고, 측정 값과 이론 값의 차이를 계산하여 기록한다. 정밀도와 정확도의 허용 기준과 동일하게 ±20% 편차(±0.1823, -17%≤%RB≤20%)를 고려하여 상한, 하한 값 설정한다. 로그변환시 등간격을 기준하여 설정한다. 끝으로, 주효과도와 교호작용도 분산분석표로 유의한 인자를 확인한다.
비교가능성 실험설계는 기존시험법과 신규시험법을 사용하여 각 3회씩 측정하여 두 시험법의 기하 평균값을 비교하였다. 측정값을 자연로그 변환하고 MINITAB21 프로그램에 입력하였다.
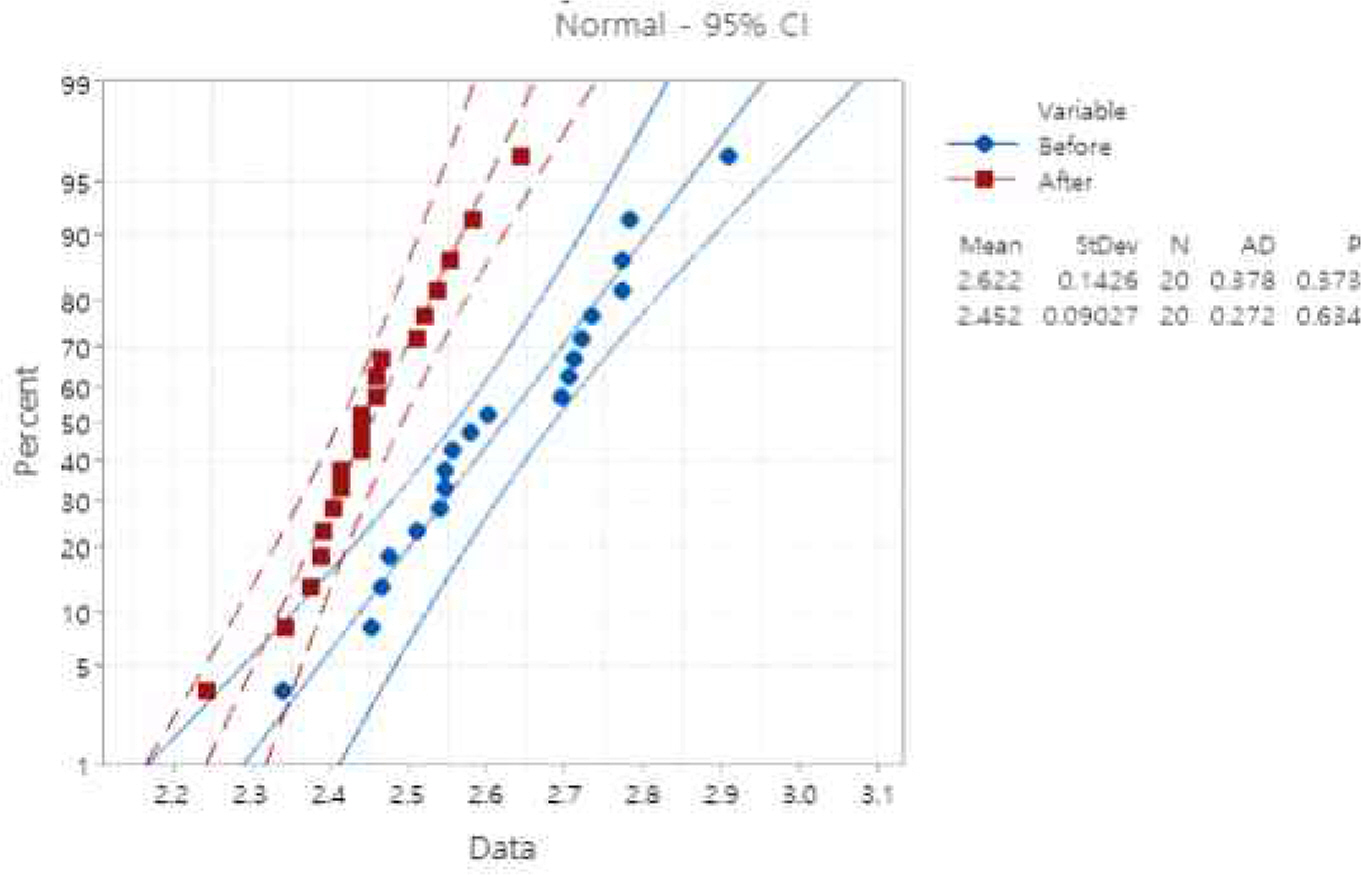
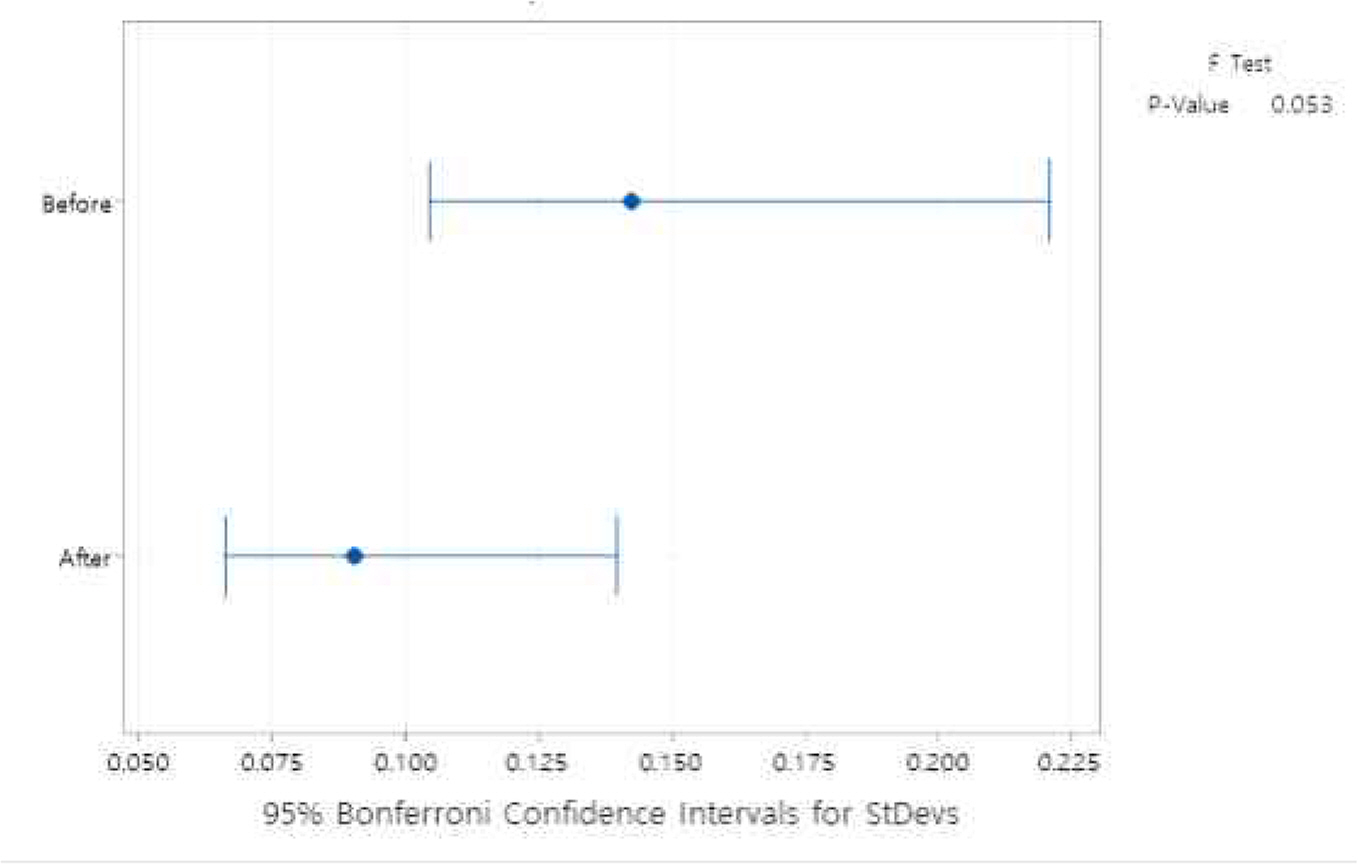
신규, 기존 시험법의 자연로그 변환 값이 통계분석의 전제조건인 정규성을 만족하며 두 그룹 간 분산이 다르지 않음을 확인하였다. Figure 4와 같이 P-값이 각각 0.373(Before: 기존 시험법), 0.634(After: 신규 시험법)로 유의수준 0.05보다 크므로 신규, 기존 시험법 결과가 정규분포를 따름을 확인하였고, Figure 5에서 P-값 0.053으로 유의수준 0.05보다 크므로 신규, 기존 시험법 분산이 다르지 않음을 확인하였다.
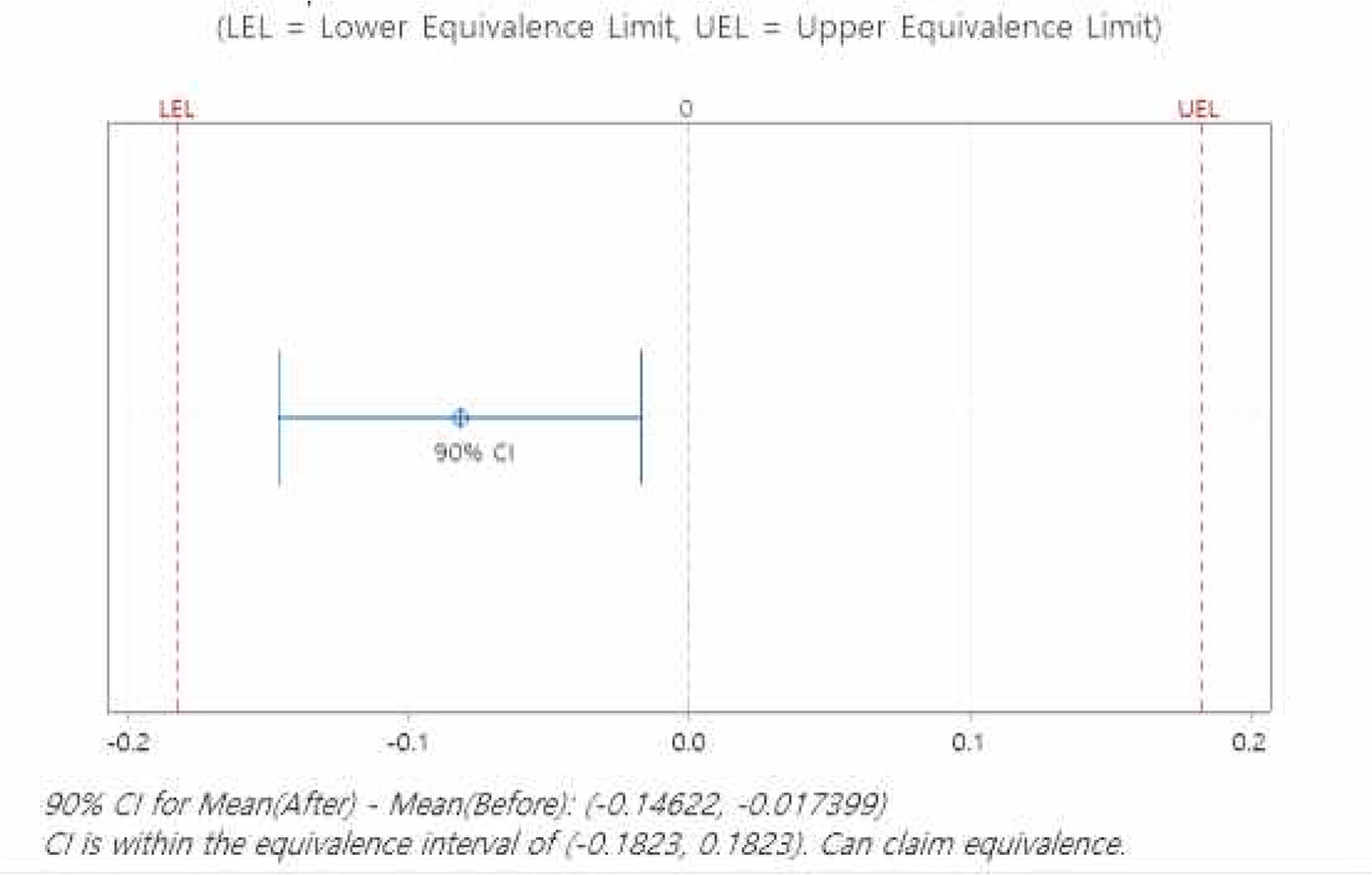
Figure 6과 같이 동등성 검정을 통해 신규, 기존 시험법의 로그 변환 값 간 차이(ln신규-ln기존)의 90% 신뢰구간이 동등성 기준 내임을 확인하였다. 하한 값은 -0.1823, 상한 값은 0.1823으로 정확도, 견고성과 같은 기준을 적용하여 설정하였다.
동등성 검정을 통해 신규, 기존 시험법의 로그 변환 값 간 차이의 90% 신뢰구간 [0.0265289, 0.0781470]이 동등성 기준 [-0.1823, 0.1823] 내 임을 확인하였다. 신규 시험법과 기존 시험법간 동등성 검정 결과, 상대적 편차의 90% 신뢰구간은 [3%, 8%]로 동등성 허용 기준 [–17%, 20%] 내로 확인되었다. 90% 신뢰 수준에서 이번 사례의 신규 시험법 분석 값이 기존 시험법 분석 값과 동등하다고 할 수 있다.
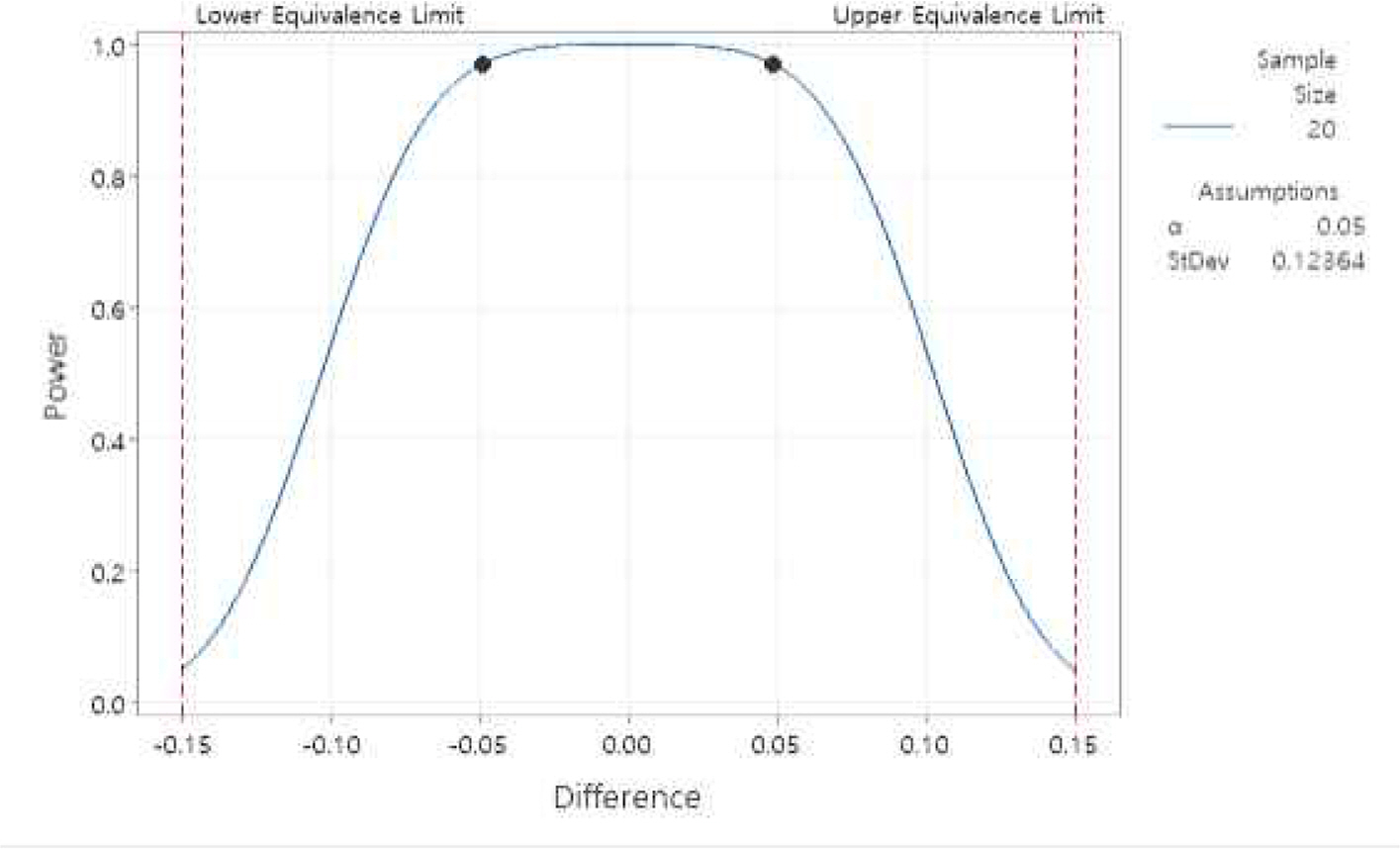
Figure 7과 같이 동등성 검정 결과의 검정력을 분석한 결과, Table 7의 MINITAB 분석결과 유의수준 5% 하에서 신규, 기존 시험법간 차이가 동등성 한계 내에 있을 때 동등하다고 판단할 확률이 약 97%임을 확인하였다. 검정력을 높이기 위해 보수적인 기준을 적용하여 표준편차는 합동표준편차가 아닌 두 그룹 간 표준편차 중 큰 값을 선택하였고, 차이 값은 점 추정치 대신 동등성 한계의 중앙값에서 먼 값을 입력하였다. 따라서 동등성 검정 결과를 신뢰할 수 있다.
신규 기존 시험법간 함량 동등성이 통계적으로 증명됨에 따라 시험법이 변경되더라도 연속적으로 품질 관리가 가능하다.
3.5 Control
이 단계에서는 정량적 성과와 정성적 성과를 정리한다. 이 연구결과 AQbD에 기반하여 시험방법 밸리데이션 시험을 수행한 보고서를 도출하였고, 위험평가를 통한 CMP 후보를 파악하고, 실험계획법을 통해 검증하였다. 기존 시험법과 신규 시험법을 통계적으로 동등함을 검증하였다. 관리전략을 수립하기 위해 시험방법 밸리데이션 결과(IP, RB)를 대입하여 시험법의 특성(Method Characterization)을 확인하고, 매 분석 배치의 표본 함량 결과를 대입하여 시험법의 시간 경과에 따른 분석 성능은 시험법의 성능지표(Capability Process Method, Cpm)로 표현할 수 있다.
USP1033의 2.5항, 3항의 계산식에 시험방법 밸리데이션 허용기준 -17%≤%RB≤20%와 결과 7.7%IP, 2.3%RB를 대입하여 시험법의 특성을 확인하였다. 시험방법 밸리데이션 허용 기준 산포와 시험방법 밸리데이션을 통해 확인된 시험법의 산포를 반복수 3을 고려하여 계산된 본 Cpm은 1.25로 적합수준으로 확인하였고, 시험법 변동에 의해 일탈이 발생할 확률은 0.017%로 낮은 수준임을 확인하였다. 시간 경과에 따른 분석 성능을 매 분석 배치마다 분석하는 품질 관리 시료의 관리도를 통해 확인한다. Table 8을 통해서 반복수가 많아질수록 Cpm성능 지수가 좋아지고, 스펙을 벗어날 확률이 현저히 작아짐을 확인할 수 있다.
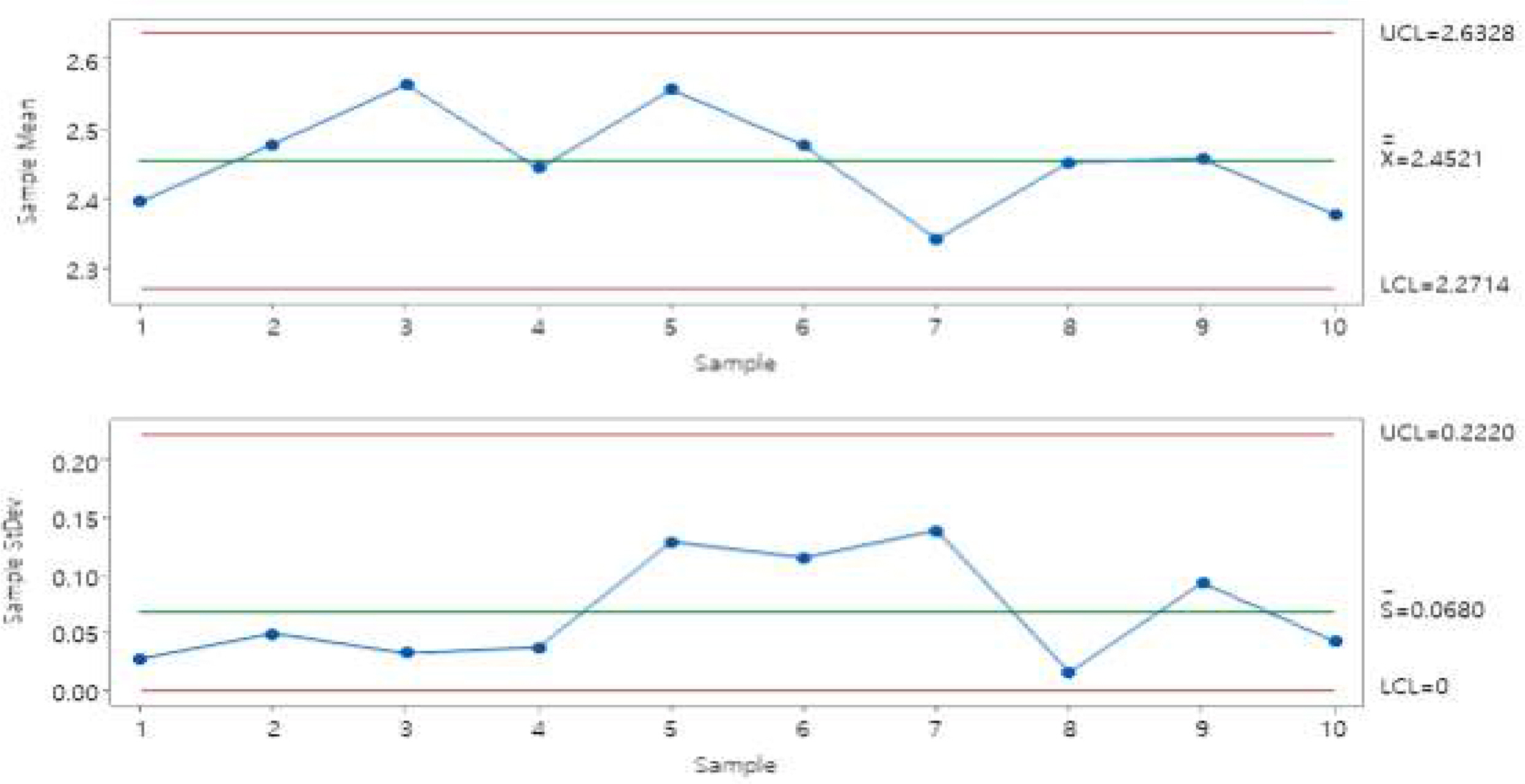
반복수가 3번이 될 때 Figure 7과 같이 XBarS 관리도로 분석 배치 간 변동과 분석 배치 내 변동 모두 3-시그마 한계선을 벗어나는 점이 없는지 확인하여 시험법 관리가 잘 되고 있다고 판단할 수 있었다. 공정능력분석을 통해 시험법의 공정능력 수준도 확인하였다. 시험방법 밸리데이션을 통해 본 시험법의 특성을 확인한 결과, Cpm=1.25, 시험법 자체 변동에 의해 OOS가 발생할 확률은 0.017% 수준으로 확인되었다. 시험방법 밸리데이션 이후 시간에 따른 시험법의 분석 성능을 품질 관리 시료를 통해 모니터링한 결과, 3-시그마 한계선을 벗어나는 점이 없고 단기공정능력과 장기공정능력의 값이 유사하여 시험법이 통계적 관리 상태에 있음을 확인하였다. 시험방법 밸리데이션에서 확인된 허용가능한 공정능력 수준으로 유지되고 있었음을 확인하였다. CS는 CMP 항목에 따라 관리전략을 수립하고 주기적인 모니터링을 실시하였다.
4. 결 론
필수의약품을 개발 및 생산할 수 있는 제약주권은 곧 국력이다. 국내의 제약회사가 글로벌 신약 회사로 발전하기 위해서는 QbD의 올바른 적용이 시급하다(Kim and Kim, 2022).
OFAT으로 시험법 밸리데이션을 수행하면 시험법에 대한 허가자료의 신뢰성이 부족하여 허가가 지연되는 리스크가 있었다. 본 연구 결과로 시험법 변경 전, 후의 D-항원 함량 동등성을 통계적인 방법으로 입증하여 시험방법 밸리데이션시 허가지연 감소를 기대할 수 있었고, 제약업계에서 빈번하게 발생되는 많은 시험법 개발 및 시험법을 검증할 때 본 논문에서 제시하는 AQbD방법으로 검증이 된다면 시험법개발 시간 단축이 가능하며, 과학적인 방법을 사용하게 됨으로써 허가 기관에 시험법 결과에 대한 신뢰를 얻을 수 있게 될 것으로 확인되었다.
QbD6시그마 프로세스로 D-항원 함량 시험법의 연구를 수행한 결과 합성 의약품과 동일한 가이드라인 ICH Q2을 적용하여 데이터 처리를 수행해 왔었는데, 본 연구를 통해 가이드라인 미국약전 USP1033을 적용하였고 데이터 전처리를 통해 데이터 정규성을 확보하는 방법을 알게 되었다. 합성 의약품의 함량과 다르게 절대적인 함량 ‘값’이 아니라 상대적인 활성 ‘비율’을 적용할 수 있음을 새롭게 알게 되었다. 생물학적 요인(Biological Factor)들의 복합적인 영향을 경험적으로 유추해왔으나, 완전요인설계법으로 시험 수행하여 생물학적 요인들의 주효과와 교호작용을 파악하게 되었다.
향후 문제해결 시 요인들의 주효과와 교호작용을 명확히 알기 위해서는 오차의 분산이 작아야 하는데, 생물검정의 측정시스템이 주로 사람이기 때문에 오차가 높아 주효과의 유의성이 감춰지는 경향이 있다. 이는 자동화기기의 도입을 통해 변동을 줄일 수 있을 것이라 판단된다. 실험 오차가 감소하여 주효과와 교호작용의 명확한 파악과 정확한 예측식 도출이 가능할 것으로 판단되나 실제 분석을 통해 확인해야 한다.
의약품 개발에 QbD를 적용할 것으로 ICH와 각국 규제기관에서 요구하고 있고, 국내 제약회사들이 글로벌 신약개발회사로 성장하기 위해서는 QbD와 AQbD를 조기에 전력화하고 체계화해서 내재화를 이룰 필요가 있다. 이에 전사적인 혁신활동으로 QbD6시그마 시스템을 도입하여 글로벌 경쟁력을 갖추는데 기여할 것으로 기대한다.